drug accumulation with multiple dosing. To do this
a loading dose, D
l
, which is larger than the main-
tenance dose, is administered. If the drug does not
show a slow disposition phase, the following
formula can be used to calculate the loading dose,
D
l
=
C
ss
,
avg
V
o
F
This same formula should be used if the drug has a
slow disposition phase and if, in addition, plasma
drug concentrations higher than the steady-state
values are to be avoided. With this loading dose,
the steady-state drug concentrations are approached
from the side of lesser concentrations, as shown in
Figure 12.5. If plasma drug concentrations higher
than the steady-state values can be safely tolerated, a
larger loading dose can be used,
D
l
=
C
ss
,
avg
V
o
F
k
el
=
C
ss
,
avg
V
F
With this dose, the steady-state drug concentrations
are approached from the side of greater concentra-
tions (Figure 12.5). V
β
is referred to as the beta
volume of distribution; it the volume that is usually
meant when referring to
the
volume of distribution.
Drug effect
When drug is administered by multiple dosing,
drug accumulates at its sites of action, eventually
achieving local steady-state concentrations. If the
kinetics at an effect site are rapid, the time it takes to
reach the steady state at the site is essentially the
same as that in the plasma. If the kinetics are slow,
the steady state will be achieved later than in the
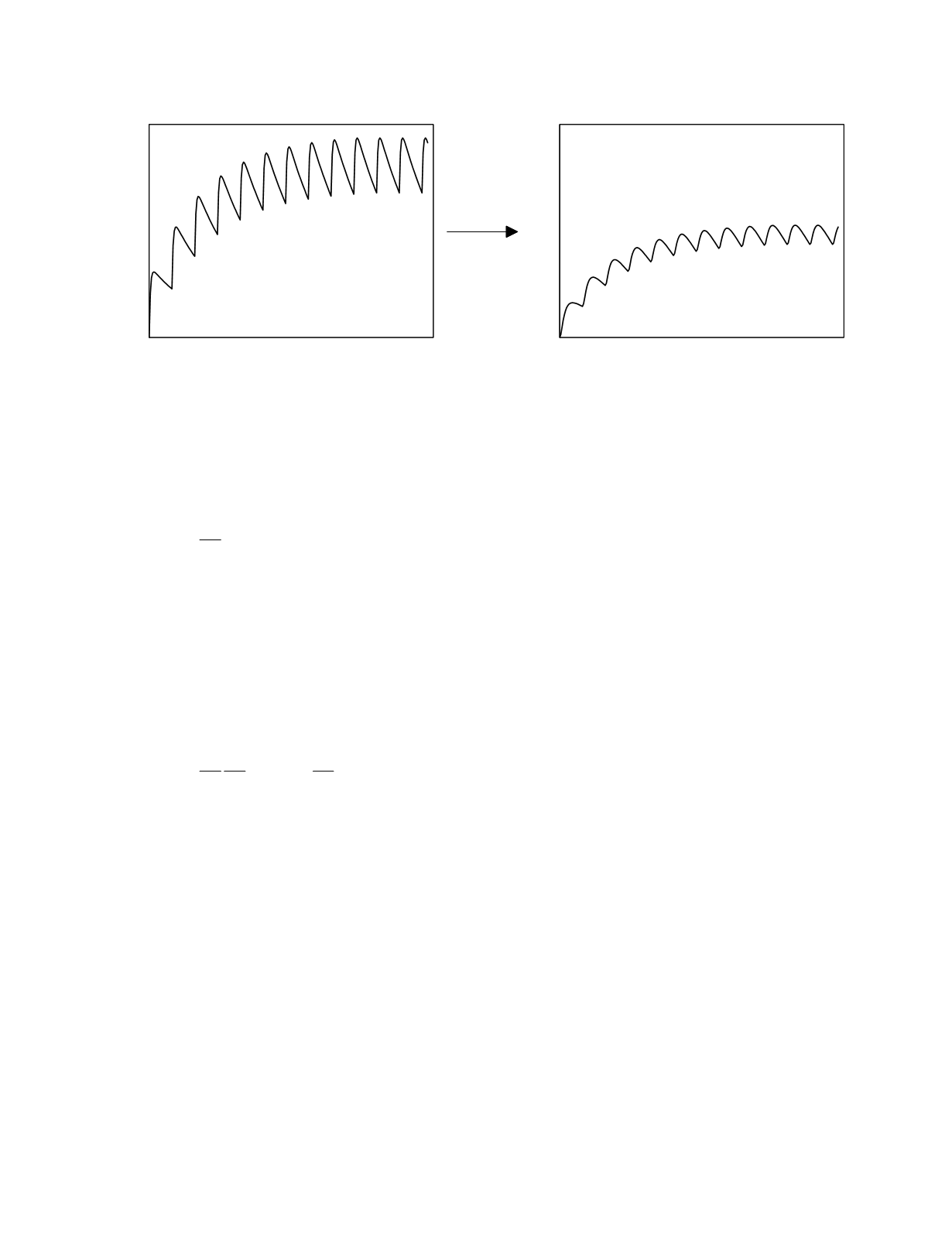
plasma. Figure 12.6 depicts the effect site drug
disposition curve for a drug with rapid effect site
kinetics. The magnitude of drug accumulation at an
effect site depends upon the plasma drug
concentrations and the effect site kinetic parameter
values. The concentrations achieved may be less
than, equal to, or greater than those in the plasma.
The kinetics of drug effect can be categorized as
instantaneous or noninstantaneous. A drug has an
instantaneous drug effect if the tissue effect evolves
rapidly and reverses rapidly. For such drugs, the
magnitude of the drug effect at any point in time is
determined by the concentration of drug present at
the site of drug action at that instant according to the
(typically sigmoidal) drug concentration-drug effect
relationship. For instance, for the drug considered
in Figure 12.6, if the kinetics of its effect were
instantaneous, the time course of the effect would
follow the time course of its effect site
concentration. There are drugs for which the magni-
tude of the drug effect is not determined by the
instantaneous drug concentration at the site of action
of the drug. This happens when there are time-
consuming intermediate steps between the local drug
effect and the observed clinical effect (delayed
effect) and when the drug effects are not rapidly
reversible so that the effect persists beyond the
period of drug exposure (cumulative effect). These
drugs show noninstantaneous drug effects.
Pharmacologic variability
For most drugs, the appropriate dose, dosing
interval, and length of treatment have been defined
by clinical studies. However, due to interindividual
variability in plasma drug kinetics and to variability
in effect site kinetics and in the drug exposure-effect
relationship, the response to a therapeutic agent
varies when the agent is administered to different
individuals. Consequently, to assure that the desired
Drug Therapy
12-5
Time
Plasma drug concentration
Time
Effect site drug concentration
Figure 12.6
Plasma and effect site drug disposition curves for an oral drug with rapid effect site kinetics.


